Machine Learning Speeds Molecular Motion Modeling
New approach yields fast, accurate model of how small organic molecules move in chemical processes.
The Science
Molecular dynamics is the study of how atoms and molecules move and interact based on traditional Newtonian physics. The method is central to many questions in modern chemistry, and computer models are a powerful tool for answering these questions. However, these models face a tradeoff between computational cost and accuracy. The alternative approach to molecular dynamics—i.e., models based on quantum mechanical physics—yields more complete results but is complex and time-consuming. Scientists have now used a machine learning technique called transfer learning to create a novel model of molecular motion. The technique is as accurate as calculations that use quantum-mechanical physics and much faster. This approach can describe a range of chemical processes and dynamics of functional organic materials.
The Impact
This is the fastest and most accurate model of small organic molecules to date. It provides a path to developing a highly accurate general-purpose model. Researchers have so far achieved these levels only with quantum chemistry calculations, which require much more computing power. This new technique promises to advance research in many fields, including drug development, reactive chemistry, and protein simulation. In nanoscience, it could advance modeling of how tiny structures grow and how molecules are arranged in self-assembled soft materials. This work could also help improve the accuracy of machine learning-based modeling in studies of metal alloys and the physics of shock and detonation.
Summary
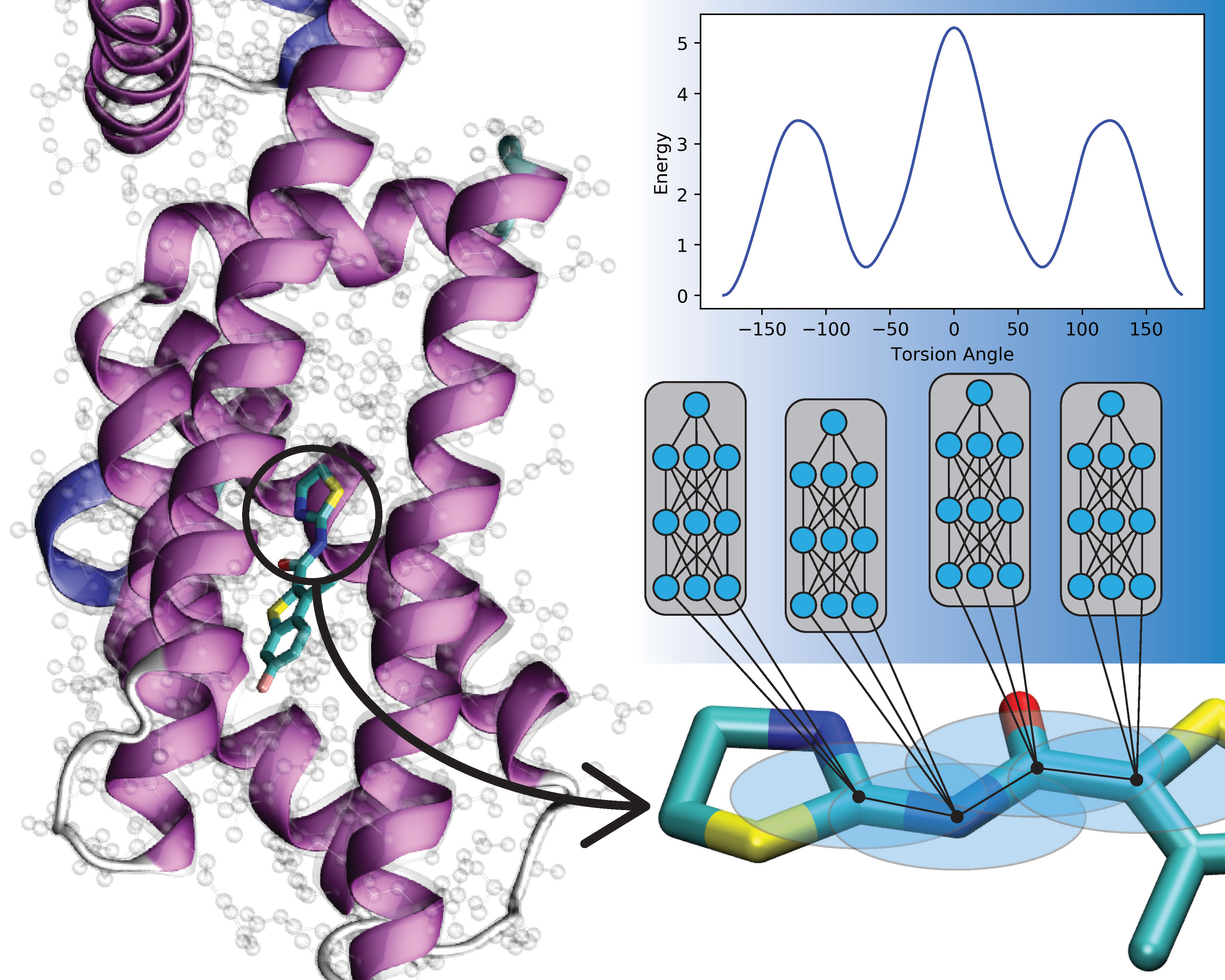
Computational models of chemical and biological systems at the atomic scale are an important tool for chemists. However, using computer simulations requires balancing between cost and accuracy. Quantum-mechanical methods are highly accurate. But they require considerable computing resources and scale poorly to large chemical and biological systems. Classical force fields are cheap and scalable, but they cannot be transferred to new systems. Machine learning methods may be the key to achieving the best of both approaches. Scientists and users from the Center for Integrated Nanotechnologies, a Department of Energy (DOE) Office of Science user facility, have used a machine learning technique called transfer learning to train ANI‑1ccx, a model of the potential energy of a molecular system. The result is a model that is accurate, transferable, and billions of times faster than the current best approach. Transfer learning methods begin with a model trained on data from one task. Researchers then retrain the model on data from a different but related task. This usually yields highly accurate predictions even when there are many gaps in the data. In this new research, researchers first trained a neural network on a large set of density functional theory data, one the most popular quantum mechanics methods for studying molecular systems. Next, they retrained the neural network on a smaller set of coupled‑cluster theory data, the gold standard for quantum mechanics calculations. The result is the best-to-date empirical model of small organic molecules, in terms of both speed and accuracy. The scientists used three test cases to benchmark the results. In each test case the machine learning approach outperformed the industry-standard methods. This model can capture a diversity of chemical processes, and it is broadly applicable to fields such as materials science, biology, and chemistry.
Contact
Sergei Tretiak
Center for Integrated Nanotechnologies, Los Alamos National Laboratory
serg@lanl.gov
Justin Smith
Los Alamos National Laboratory
just@lanl.gov
Funding
This research was a joint study by investigators at Los Alamos National Laboratory (LANL), the University of Florida, Jackson State University, and the University of North Carolina. The authors acknowledge support of the Department of Energy through the LANL Laboratory Directed Research and Development program. The research was performed, in part, at the Center for Integrated Nanotechnologies, a DOE Office of Science user facility and with computational resources from the LANL Institutional Computing program. Research was supported by the National Science Foundation; a University of Florida graduate student fellowship, the LANL Center for Non-linear Studies; the Department of Defense Office of Naval Research, and the Eshelman Institute for Innovation. Also acknowledged was a hardware donation from NVIDIA Corporation and the Open Science Grid (funded by the National Science Foundation and the DOE Office of Science).
Publications
Smith, J.S. et al., “Approaching coupled cluster accuracy with a general-purpose neural network potential through transfer learning.” Nature Communications 10, 2903 (2019). [DOI: 10.1038/s41467-019-10827-4]
Related Links
Los Alamos National Laboratory News Release: Researchers cast neural nets to simulate molecular motion
Highlight Categories
Performer: University , DOE Laboratory , SC User Facilities , BES User Facilities , CINT
Additional: Collaborations , Non-DOE Interagency Collaboration